


Un nuevo método computacional captura la dinámica molecular tras la excitación ultrarrápida de varios estados coherentes
06.03.2026
 |
|
|
- Investigadores liderados por el profesor Fernando Martín capturan cómo interactúan los electrones y los núcleos tras la excitación de luz attosegunda de una forma numéricamente económica.
- Este enfoque, único a la hora de capturar el efecto de la excitación inicial de varios estados, abre nuevas oportunidades para comprender el movimiento de la carga en moléculas complejas.
| Tweet |
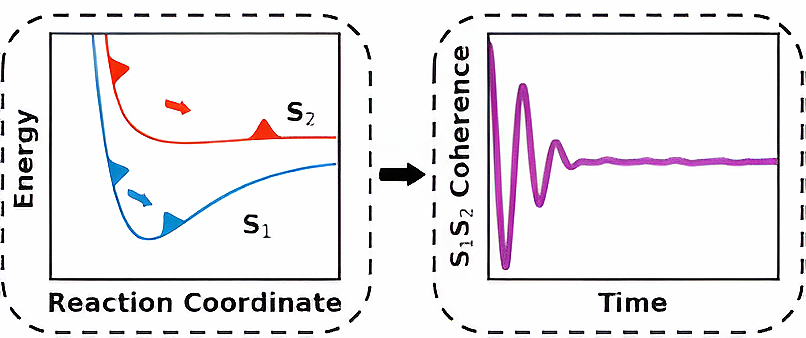
Madrid, 6 de marzo, 2026. Los científicos han desarrollado un nuevo y potente método computacional que ayuda a explicar lo que ocurre dentro de las moléculas cuando son golpeadas por ráfagas de luz extremadamente cortas. Los modernos pulsos láser de attosegundos y femtosegundos pueden desencadenar varios estados electrónicos en una molécula a la vez, creando una delicada mezcla cuántica conocida como coherencia electrónica. Comprender cómo evoluciona esta coherencia —y cómo se interrumpe cuando los núcleos comienzan a moverse— ha sido un desafío desde hace mucho tiempo, lo que limita nuestra capacidad para interpretar los experimentos de espectroscopia ultrarrápida en moléculas complejas.
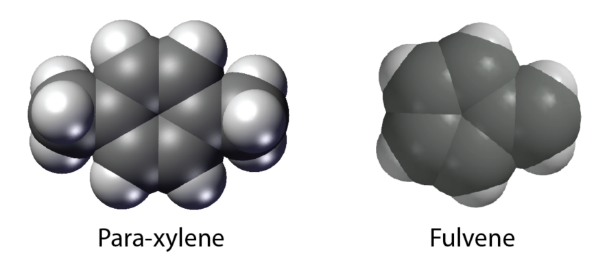
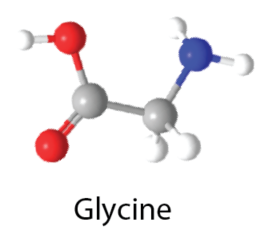
El nuevo método, denominado Trajectory Surface Hopping with Projected Forces and Momenta (TSH-PFM), proporciona una forma eficaz de simular el movimiento entrelazado de los electrones y los núcleos durante estos procesos ultrarrápidos. A diferencia de los enfoques anteriores, el TSH-PFM captura la excitación de varios estados coherentes. El método tiene en cuenta efectos cuánticos clave, como la pérdida y la regeneración de la coherencia electrónica y el complejo comportamiento que surge cerca de sitios moleculares especiales conocidos como intersecciones cónicas. Los investigadores validaron el método reproduciendo con éxito resultados cuánticos conocidos para varias moléculas de referencia, entre ellas el paraxileno y el fulveno, mientras trabajaban en dimensionalidad completa.Para demostrar el impacto más amplio del método, el equipo lo aplicó a la glicina, el aminoácido más simple. Las simulaciones revelaron que las coherencias electrónicas iniciales pueden remodelar drásticamente la forma en que se distribuye la carga eléctrica a través de la molécula en los primeros momentos después de la excitación.
Para obtener un modelo teórico detallado de la dinámica tras la excitación de una molécula, es necesario tener en cuenta la sutil interacción entre los electrones y los núcleos, que en última instancia subyace a la formación de enlaces químicos. En su último trabajo, investigadores de la Universidad Autónoma de Madrid y del instituto IMDEA Nanociencia han logrado capturar los efectos electromagnéticos y cuánticos subyacentes de una forma numéricamente económica, allanando el camino para interpretar futuros experimentos en química, biología y ciencia de los materiales que investiguen moléculas complejas en sus escalas de tiempo más rápidas.
El trabajo es el resultado del proyecto europeo TOMATTO, financiado por una subvención ERC Synergy (G.A. 951224). TomATTO tiene como objetivo capturar la dinámica ultrarrápida de los electrones con el fin de mejorar la eficiencia de la conversión de la energía solar.
Referencia:
J. Chem. Theory Comput. 2025, 21, 21, 10645–10668
https://doi.org/10.1021/acs.jctc.5c00531
![]() Link al repositorio: https://hdl.handle.net/20.500.12614/4153
Link al repositorio: https://hdl.handle.net/20.500.12614/4153
Contacto:
Oficina de Divulgación y Comunicación en IMDEA Nanociencia
divulgacion.nanociencia [at]imdea.org![]()
![]()
![]()
![]()
![]()
Fuente: IMDEA Nanociencia.
El Instituto IMDEA Nanociencia es un centro de investigación interdisciplinar en Madrid dedicado a la exploración de la nanociencia y el desarrollo de aplicaciones de la nanotecnología en relación con industrias innovadoras. IMDEA Nanociencia es un centro de Excelencia Severo Ochoa desde 2017, máximo reconocimiento a la excelencia investigadora a nivel nacional.